

在2026生物医药产业合作大会关注的前沿肿瘤研究领域中,DNA修复机制再次成为焦点。近日,发表于《Science》的最新研究揭示了癌细胞在BRCA2缺陷条件下仍能存活的深层原因:一个名为FIGNL1的蛋白分子,或许才是癌细胞“难杀”的关键。

图片来源:学术经纬
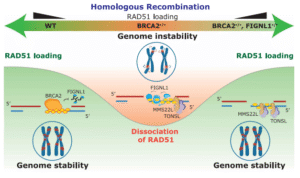
细胞每天都在经历数以万计的DNA损伤事件,而同源重组修复(HR)是最关键的修复通路之一。在这一过程中,BRCA2与RAD51两种蛋白发挥核心作用:BRCA2帮助RAD51结合DNA断裂位点,从而触发修复程序。当BRCA2功能缺失时,RAD51无法有效装载,导致DNA断裂得不到修复,也解释了为何BRCA2突变与乳腺癌、卵巢癌和前列腺癌的高发密切相关。
然而,科学家们注意到一个异常现象:部分BRCA2缺陷的癌细胞仍能部分恢复修复功能。最新研究首次揭示了其中的机制——问题不只是RAD51“装不上去”,而是被过度移除。研究发现,FIGNL1蛋白会以“分子马达”的方式利用ATP能量,将已结合的RAD51主动从DNA上剥离,从而削弱修复通路。
当研究者在人类及小鼠的BRCA2缺陷细胞中敲除FIGNL1后,RAD51重新回到了DNA断裂位点,DNA修复效率显著恢复。同源重组功能的重建显著提高了基因组稳定性。更令人惊讶的是,在通常无法存活的BRCA2缺陷型小鼠胚胎干细胞中,同时去除FIGNL1后,细胞竟恢复了活力。
图片来源:学术经纬
研究进一步揭示,FIGNL1不仅与RAD51直接结合,还以六聚体结构形式存在,具备独特的RAD51结合域。作为ATP酶,FIGNL1可利用ATP水解能量将RAD51丝状结构从单链DNA上解离。BRCA2的正常功能则能对抗FIGNL1的移除活性,保护RAD51稳定性,确保修复过程顺利进行。 一旦BRCA2缺失,FIGNL1活性失控,导致修复系统彻底崩溃。
这一发现不仅重塑了科学界对同源重组的理解——它不再只是BRCA2和RAD51之间的“双人舞”,而是由三者共同调控的动态平衡系统:BRCA2促进结合、RAD51执行修复、FIGNL1负责移除。
更具临床意义的是,这一机制为肿瘤耐药性提供了关键解释。以PARP抑制剂为代表的靶向疗法,原理在于同时切断肿瘤的单链与双链修复通路,使癌细胞“无路可走”。但随着治疗进行,部分肿瘤逐渐产生耐药。该研究提示,癌细胞可能通过抑制FIGNL1功能,重新恢复RAD51装载,从而“修复自愈”,逃避药物杀伤。
研究团队指出,联合靶向PARP与FIGNL1可能是下一步的精准治疗方向,可望彻底破坏癌细胞的DNA修复网络,为耐药性肿瘤提供新疗法思路。
2026生物医药产业合作大会关注到,这一成果不仅深化了对肿瘤DNA修复网络的理解,也为未来的精准医疗和药物联合策略提供了新的科学依据,或将在未来肿瘤治疗与药物开发投资领域引发持续热议。
文章来源:学术经纬
